使用luigi库进行工作流管理的整个exome分析管道的实现。
wespipeline的Python项目详细描述
WESPIPEline
使用用于工作流管理的Luigi <https://github.com/spotify/luigi/>实现整个exome分析管道。
。图::https://raw.githubusercontent.com/janchorizo/wespipeline/master/docs/steps.png :alt:steps徽标 :对齐:居中
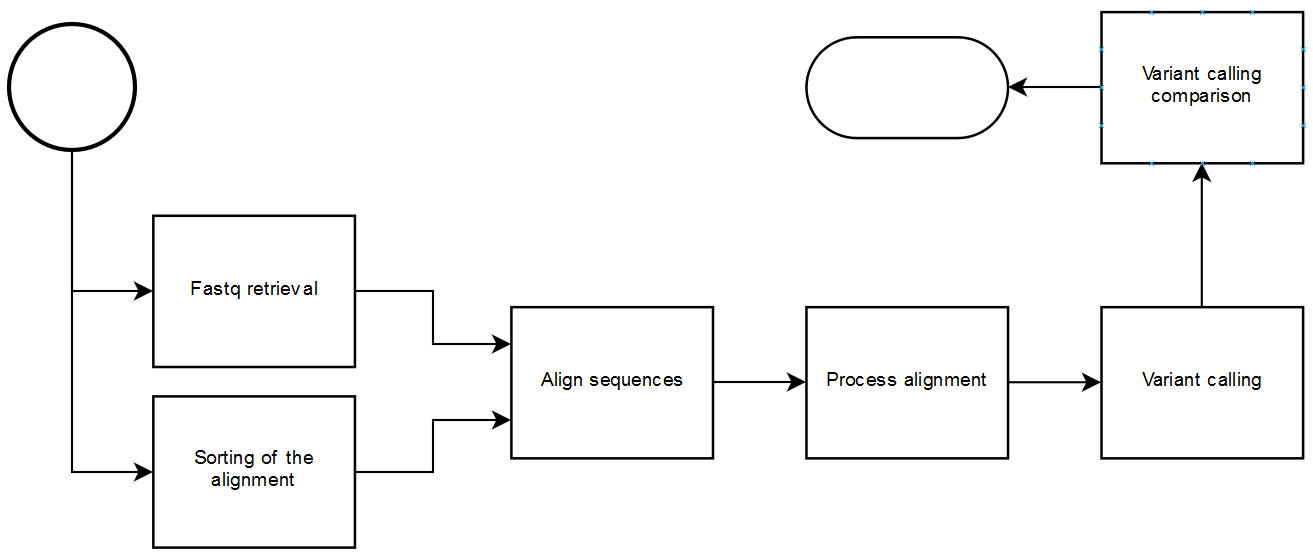{kind=link}
此包提供执行部分或完整变量调用的任务的实现 分析具有工作流管理器的优点:依赖关系解析、执行计划器, 模块化、监控和历史性。
最新版本的文档由readthedocs <https://wespipeline.readthedocs.io/en/latest/>主持
安装
^^^^^^^^^^^^
WESPIPELINE可通过PIP、CONDA和手动安装获得。从包存储库安装
pip3 install wespipelineconda install -c jancho wespipeline,或者下载项目并从源代码生成:
git clone https://github.com/Janchorizo/wespipeline.git && cd wespipeline && python3 setup.py install。
注意,根据执行的步骤和 为这些设置的参数。下面引用了所有可能的内容,可以通过anaconda发行版下载:
- 安全检索:SRA工具包,FastQC
- 参考基因组检索:无需依赖性
- 合流对齐:bwa
- 对齐处理:bwa samtools,
- 变量调用:freebayes、varscan、gatk、deepvariant
- 变型呼叫评估:VCF工具
除了依赖项之外,conda还可以用于安装wespipeline包。一个例子 安装miniconda发行版、包和依赖项是:
。代码块::bash
wgethttps://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh-o~/miniconda.sh公司
bash~/miniconda.sh-b-p$home/miniconda
export path=“$home/miniconda/bin:$path”
源$HOME/miniconda/bin/activate&;
conda config—添加频道bioconda&;
conda config—添加频道conda forge&;
conda config—添加频道jancho&;
conda安装-y samtools&;br/>
Conda安装-y bwa&;br/>
conda安装-y picard&;br/>
Conda安装-Y鸭嘴兽变型
Conda安装-y varscan&;br/>
Conda安装-Y Freebayes&;br/>
conda安装-y fastqc&;br/>
conda安装-y sra tools&;
Conda安装-Y WESPIPEline
rm~/miniconda.sh
入门 ^^^^^^^^^^^^^^^
安装或下载软件包将为 分析,每个分析都可以以类似于其他luigi任务的方式执行。
六个步骤中的每一个都有一个更高级别的任务,可以以类似的方式安排 其他Luigi任务:
。代码块::bash
python3 -m luigi --module wespipeline.<module> <Taskname> --<Taskname>-param value
使用NCBI登录号下载序列。
。代码块::bash
python3 -m luigi --module wespipeline.fastq FastqRetrieval \
--FastqRetrieval-paired-end true \
--FastqRetrieval-accession-number SRR9209557 \
--FastqRetrieval-create-report true
或外部url。
。代码块::bash
python3 -m luigi --module wespipeline.fastq FastqRetrieval \
--FastqRetrieval-paired-end true \
--FastqRetrieval-compressed false \
--FastqRetrieval-accession-number SRR9209557 \
--FastqRetrieval-create-report true
下载参考基因组并使用fastqc创建报告。
。代码块::bash
python3.6 -m luigi --module tasks.reference ReferenceRetrieval
--workers 3 \
--ReferenceGenome-ref-url ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit \
--ReferenceGenome-from2bit True \
--GlobalParams-base-dir ./tfm_experiment \
--GlobalParams-log-dir .logs \
--GlobalParams-exp-name hg19
或者运行整个分析,为每个步骤指定参数。
。代码块::bash
python3 -m luigi --module tasks.vcf VariantCalling
--workers 3
--VariantCalling-use-platypus true
--VariantCalling-use-freebayes true
--VariantCalling-use-samtools false
--VariantCalling-use-gatk false
--VariantCalling-use-deepcalling false
--AlignProcessing-cpus 6
--FastqAlign-cpus 6
--FastqAlign-create-report True
--GetFastq-gz-compressed True
--GetFastq-fastq1-url ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/Garvan_NA12878_HG001_HiSeq_Exome/NIST7035_TAAGGCGA_L001_R1_001.fastq.gz
--GetFastq-fastq2-url ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/Garvan_NA12878_HG001_HiSeq_Exome/NIST7035_TAAGGCGA_L001_R2_001.fastq.gz
--GetFastq-from-ebi False
--GetFastq-paired-end True
--ReferenceGenomeRetrieval-ref-url ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit --ReferenceGenomeRetrieval-from2bit True
--GlobalParams-base-dir ./tfm_experiment
--GlobalParams-log-dir .logs
--GlobalParams-exp-name hg19
执行的任务 ^^^^^^^^^^^^^^^^^
+——+———————————————————————————————————————————————--+ |模块任务| +=================+============================+ |参考文献| +——+———————————————————————————————————————————————--+ | fastq fastqretrieval公司| +——+———————————————————————————————————————————————--+ |对齐快速对齐| +——+———————————————————————————————————————————————--+ |处理对齐快速处理| +——+———————————————————————————————————————————————--+ |变数调用变数调用| +——+———————————————————————————————————————————————--+ |处理对齐差异处理| +???+???+
致谢 ^^^^^^^^^^^^^^^^
特别感谢路易斯·安东尼奥·米格尔·金塔莱斯教授的指导和帮助。在 开发这个项目。
欢迎加入QQ群-->: 979659372
